Details
-
Bug
-
Status: Resolved
-
 Major
Major
-
Resolution: Cannot Reproduce
-
Simulator 1.0.3 (API 1.12)
-
None
Description
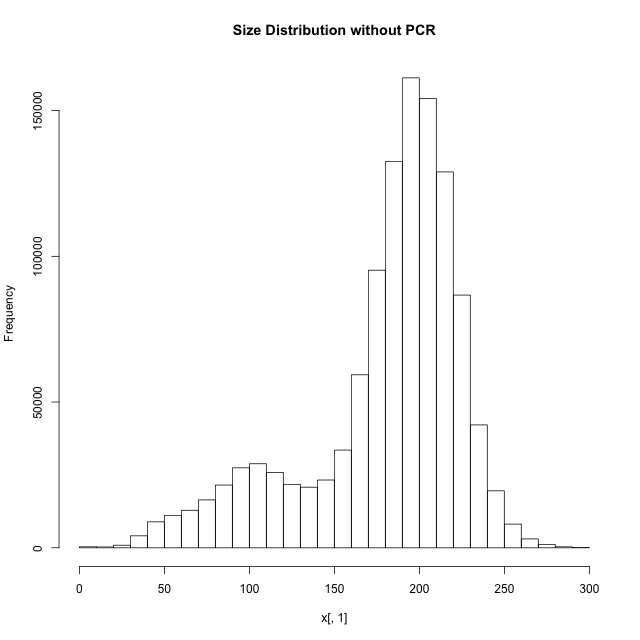
When I continue to PCR amplification of these 2 million fragments, it gives me 55 million fragments and then the fastq file. Now, when I map them using tophat to my genome, only 30% of the reads mapped. When I tried to find the error by mapping using BWA, I found that the inner distance between the reads were around 500-1200. Again, how is this possible? BWA is not a spliced read aligner and it gives me inner distance this high when my fragments are supposedly between 181 and 300 (most of them anyways, assuming there would be more reads from non-spliced regions than splice junctions). When you mean fragment size, does it include adapters? If so, assuming 35bp on both side the effective insert size is 300-70 = 230 bp. If you mean insert size as fragment, then its 300. Eitherways, the inner distance between the two pairs must be between (230 - 76*2)=78 and (300-76*2)=148 on an average. But when I calculated from my script to obtain mean and sd of inner distance, mean=360 and sd=760 (after mapping). Is there an explanation for this? Or am I understanding something totally wrong? This is not possible unless almost all such reads are generated over splice junctions… isn't it?
BWA output:
[infer_isize] (25, 50, 75) percentile: (96, 212, 732) [infer_isize] inferred external isize from 133773 pairs: 406.571 +/- 449.596 [infer_isize] inferred maximum insert size: 3199 (6.21 sigma) ... [infer_isize] (25, 50, 75) percentile: (234, 705, 1613) [infer_isize] inferred external isize from 141160 pairs: 962.510 +/- 918.874 [infer_isize] inferred maximum insert size: 6568 (6.10 sigma)